21-Dec-2023
Escherichia coli is one of the most common and well-documented bacteria. Adherent-Invasive E. coli (AIEC) is a subclass often found in the human gastrointestinal tract, but their low abundance, plus a healthy immune response, makes them little threat. However, AIEC is highly prevalent in those suffering from Crohn’s disease – any deeper understanding of the method of cell invasion and survival could be crucial to the development of drugs to treat the disease.
IbeA is a protein which had only been identified in pathogenic E. coli, originally in the K1 strain, where it was shown to be involving in the crossing of the blood-brain barrier. It has also been shown to be important for host cell invasion in the gut. A team from University of Montpellier accessed structural biology technologies at IBS Grenoble, (Instruct-FR2), utilising a combination of size exclusion chromatography (SEC), mass spectrometry (MS), 3D modelling and various other techniques to investigate the structure, binding target(s), and binding mechanism of IbeA.
Laure Yatime of University of Montpellier said, “This is the second time that we get the opportunity to use the MS platform at IBS Grenoble through Instruct-funded access. The combination of MS techniques we used for the IbeA project was instrumental in elucidating the nature and biological function of this protein that is very important for the AIEC bacteria to survive within host cells and escape immune control inside the gut."
A soluble form of IbeA was expressed in E. coli cytosol, rather than membrane-bound as had been reported in previous studies. SEC revealed two peaks, indicating main and secondary forms of the protein that were both larger than simple monomers. The oligomerisation state was then analysed by native MS, which indicated that both IbeA forms are homodimeric. With direct infusion MS, a small molecule was found to have bound to IbeA – the likelihood was that this was a flavin cofactor, and was observed then to be flavin adenine dinucleotide (FAD).
With three peaks displayed in native MS, the conclusion was that IbeA existed as three separate forms in this context: the standard apo IbeA homodimers, the IbeA homodimers bound to one molecule of FAD, and those bound to two molecules of IbeA. This indicates that IbeA is a flavoprotein, which can bind up to two FAD molecules per homodimer in reducing conditions. Reducing conditions also prevent the prevalence of large, unstable oligomers and allow additional binding of exogenous FAD in a dose-dependent manner.
To understand the structure of IbeA, prediction software AlphaFold was used to examine its tertiary structure. The model found that IbeA has two distinct domains with an intermediate hinge region. Domain A forms a classic FAD binding domain; Domain B is less structured. The model also found that IbeA contains ten cysteine residues, six of which are located on its surface – this may indicate why clustering occurs and the large oligomeric structures form in non-reducing conditions.
Comparing this model with those in the PDB demonstrates that IbeA is similar to FAD-dependent oxidoreductases, most specifically with geranylgeranyl reductases (GGR). Using AlphaFold to compare these, shows strong similarity, particularly in the Domain A FAD-binding region. Partial similarity was also found in Domain B. The specific function of this second domain remains unclear.
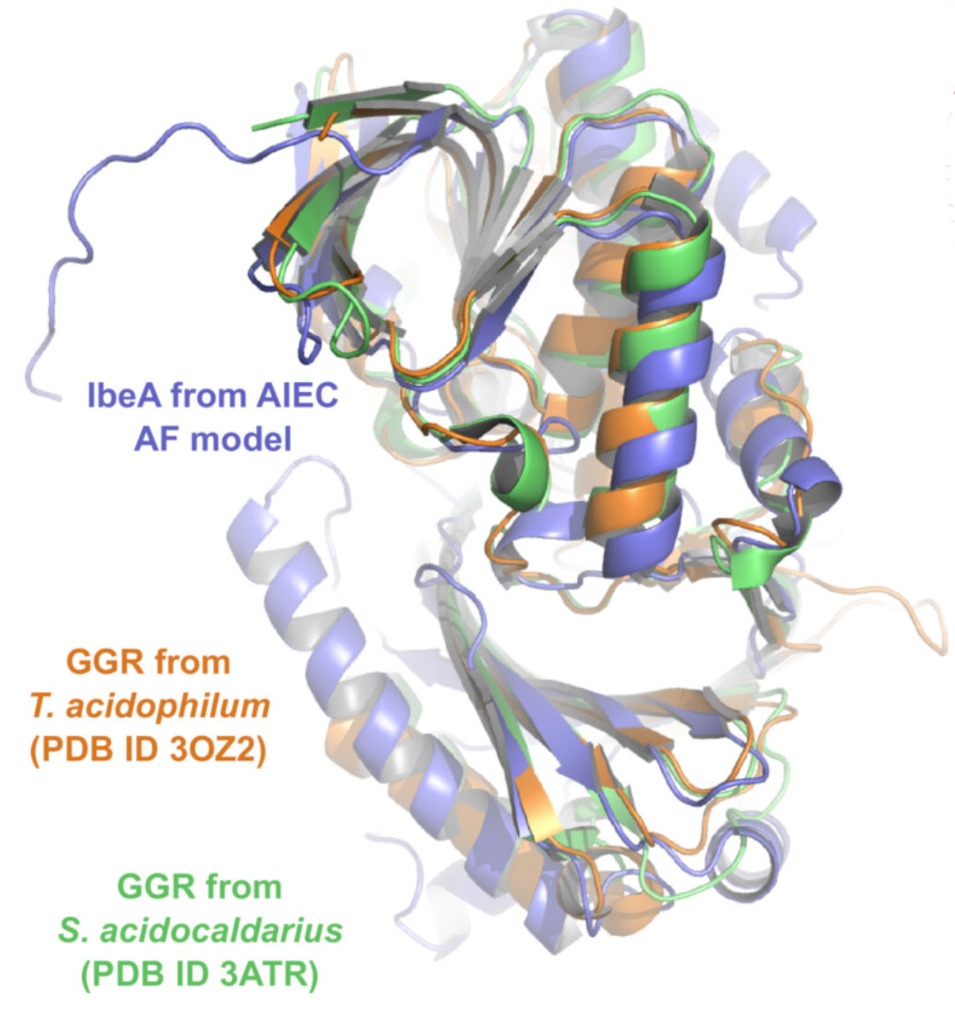
IbeA (purple) overlain with GGR structures (orange and green) using AlphaFold prediction software.
Using AlphaFold to explore the structure of IbeA as a homodimer, finds that it contacts between both A domains, and both B domains, exhibiting 2-fold symmetry. There is no strong change to its conformational or oligomeric state when bound to FAD, although the reaching of this stable state is faster when IbeA is bound to FAD.
Through the use of native MS at IBS Grenoble (PID 17773), the team was able to identify the ligand of IbeA and how IbeA bound to it, and gain insight into IbeA’s structure and how this protein might help facilitate cell invasion and survival. For such a key E. coli protein, this initial study is significant in understanding its function, and can be a crucial step in developing potential drugs against the bacteria.
On the access procedure, Laure said, "The whole process went on very smoothly from proposal writing to platform access and we could have all our samples tested in a relatively short time frame. We would certainly not have been able to perform such an integrative study without the Instruct infrastructures”.
