We have applied some changes to ARIA and are monitoring stability, but you can continue to work as normal. We appreciate your understanding.
23-Jul-2021
More than 350,000 structure predictions will be made freely available to the structural biology community through the AlphaFold Database.
Predicting the 3D structure of a protein based on its amino acid sequence has been an ongoing challenge for structural biologists since its introduction in the 1970’s. However, throughout this period it was determined that there was a strong correlation between protein amino acid sequences, and their experimentally determined structures.
DeepMind is an artificial intelligence (AI) organisation, working across a wide range of disciplines. In 2020, its AI AlphaFold system was recognised as the best method for predicting 3D protein structures, significantly outperforming its contemporaries, achieving a median of 0.96 Å compared to 2.83 Å for the next-best method. It consistently predicted 3D structures very similar to the experimentally determined structures in the 14th round of the biennial CASP (Critical Assessment of protein Structure Prediction) experiment - a regular objective test of protein structure prediction methods.
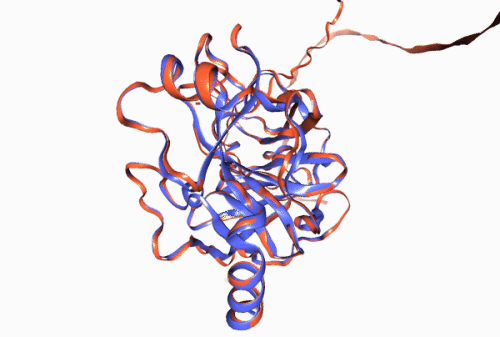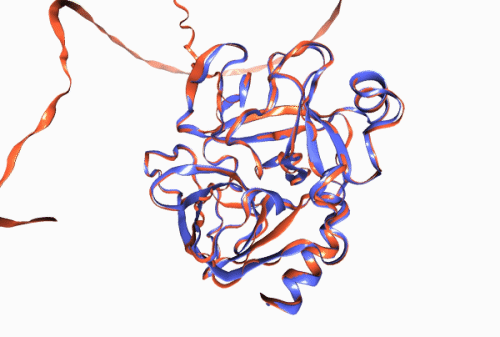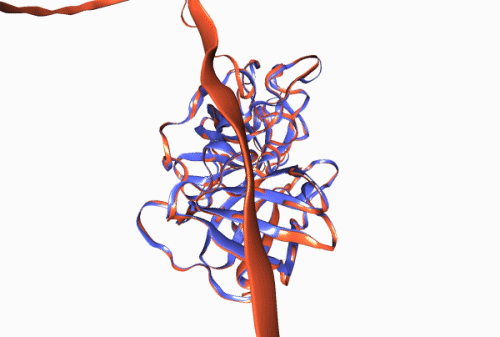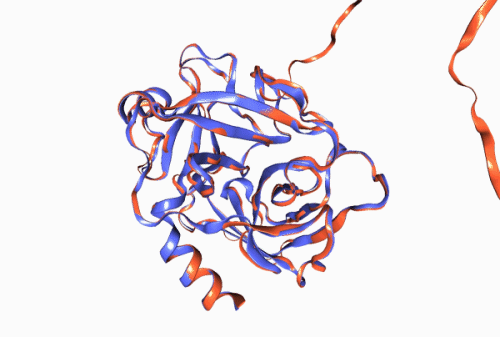
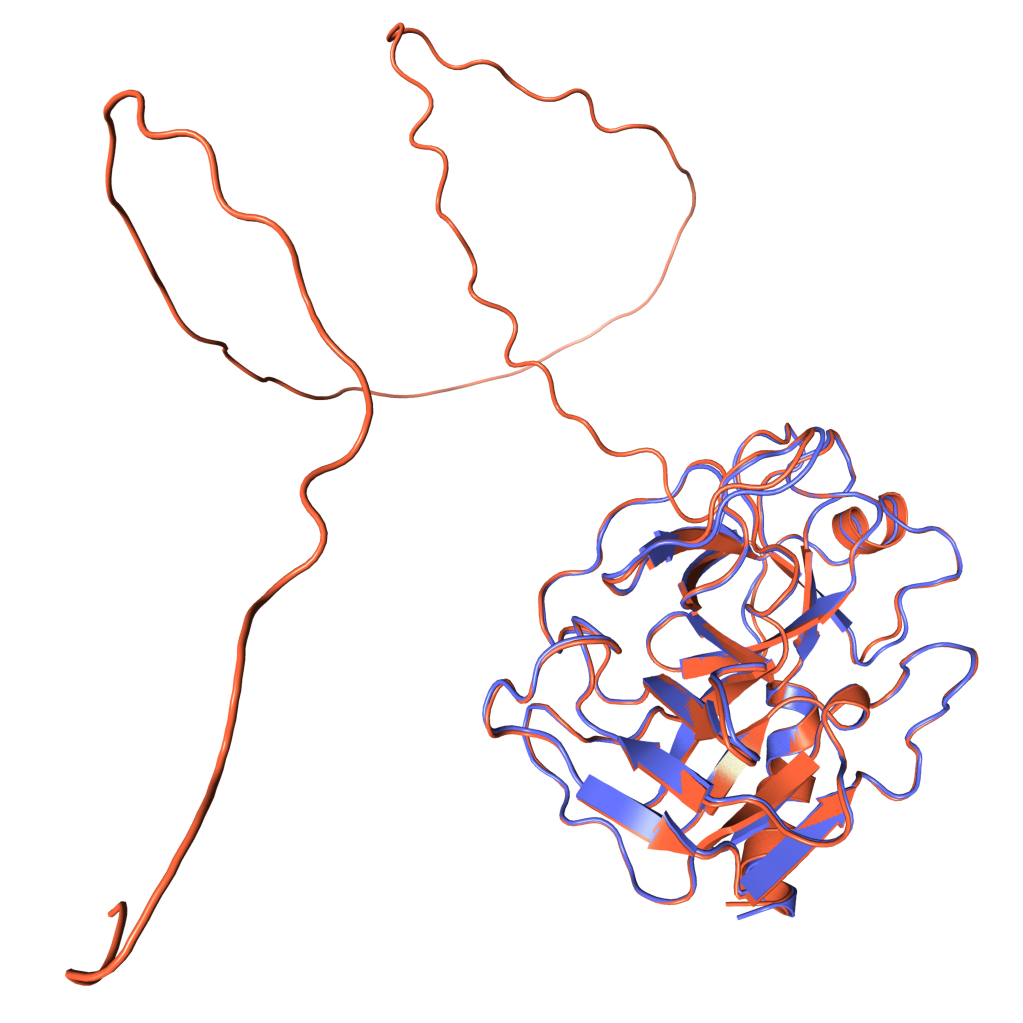
Figure 1. Experimentally derived (blue) and AlphaFold prediction (red) of Trypsin 3 protein structure.
Hundreds of thousands of structure predictions will be made freely available to the community through the AlphaFold Database (DB). This resource, partnered by DeepMind and EMBL European Bioinformatics Institute (EMBL-EBI) is based on the rich stock of experimental structures archived in the Protein Data Bank, and a variety of other databases – the total number of predicted structures at launch is over 350,000, aiming to increase to an estimated 130 million 3D models.
EMBL is a member of Instruct-ERIC, with three facilities offering services to Instruct researchers: Grenoble, Hamburg and Heidelberg.
Professor Sir David Stuart FRS, Director of Instruct-ERIC, commented, “EMBL has an ongoing aim to promote open research data and open science throughout the structural biology community – this ground-breaking data resource is an incredibly effective way of doing just that.
“The initial benefit for researchers will be clear to see – improved structure studies and hypotheses in the near future. But the potential long-term impact of such a step change in protein structure prediction could be extremely significant, in drug discovery, protein dynamics, even into biology as a whole.”
The initial release of the resource provides structure predictions for most of the proteins in the human proteome as well as for the proteomes of 20 other species of significant biological or medical interest.
For more information on AlphaFold DB, visit the EMBL website.
Instruct-ERIC centres have also begun providing additional services using Alphafold, find out more below: