Featuring expert speakers from Instruct Centres across Europe, Instruct-ERIC Webinar Series: Structure Meets Function highlights some of the latest developments in structural biology, demonstrating how integrative methods are enabling scientists to decipher the mechanisms that underpin health and disease.
Watch the previous webinars in the series here.
The 16th webinar in the series will be hosted by Instruct Israel on 8 February 2022, 11:00 - 12:00 CET. Register for the webinar here.

Webinar Moderator: Michal Sharon, Weizmann Institute of Science
Talk 1: SARS-CoV-2 Omicron prediction and antiviral drug design are enabled by RBD in vitro evolution
Speaker: Gideon Schreiber
Affiliation: Weizmann Institute of Science
Abstract: SARS-CoV-2 variants of interest and concern will continue to emerge for the duration of the COVID-19 pandemic. To map mutations in the receptor-binding domain (RBD) of the spike protein that affect binding to ACE2, the receptor for SARS-CoV-2, we applied in vitro evolution to affinity-mature the RBD. Multiple rounds of random mutagenic libraries of the RBD were sorted against decreasing concentrations of ACE2, resulting in the selection of higher affinity RBD binders. We found that mutations present in more transmissible viruses (S477N, E484K/A, Q498R and N501Y) were preferentially selected in our high-throughput screen. Evolved RBD mutants include prominently the amino acid substitutions found in the RBDs of Alpha, Beta, Gamma and the recently emerging Omicron variant. Of special interest is Q498R, which is epistatic to N501Y and increased binding affinity by 30-fold. This mutation allowed for the RBD of Omicron to rearrange in a way that makes it stealth to the presence previously infected patient sera, monoclonal antibodies and the most commonly used vaccines. Further in vitro evolution increased the binding affinity of the RBD to pM (termed RBD-62), which is a potent drug inhibiting infection of SARS-CoV-2, independent of the virus variant. This was shown by inhibition of infection in cell cultures and in non-human primates. A 2.9 Å cryo-electron microscopy structure of the high-affinity complex of RBD-62 and ACE2, including all rapidly spreading mutations, provides a structural basis for RBD-62 activity.
References:
1. Dejnirattisai W et al. (2022) SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell DOI:https://doi.org/10.1016/j.cell.2021.12.046
2. Zahradník J et al. (2021) SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat Microbiol 6(9):1188–1198.
Talk 2: AlphaFold and protein optimisation
Speaker: Shiran Barber-Zucker
Affiliation: Weizmann Institute of Science
Abstract: Reliable and completely computational protein optimisation:
Our lab has developed reliable methods for optimizing a variety of functional properties in proteins. Proteins designed using our tools exhibit high thermal stability, heterologous expression levels, catalytic rate, and binding affinity. Our methods, however, demand atomically accurate structures, and until recently, this has meant that they depended on experimental structures. The advent of high-accuracy AI-based modelling software, such as AlphaFold, opens the way to optimize proteins for which no experimental structure is available. We demonstrate how AI-based models can be used as reliable starting points for design calculations. We targeted a family of fungal enzymes, called versatile peroxidases, for stability design. The designed variants could be functionally expressed in yeast cells (whereas the wild type proteins could not) and demonstrated a wide spectrum of functional properties. Versatile peroxidases are critical in the biodegradation of lignin and the designs may have important uses in commercial biofuel production. More broadly, our method, which is completely automated and available online, extends the scope of computational protein optimization essentially to any natural protein.
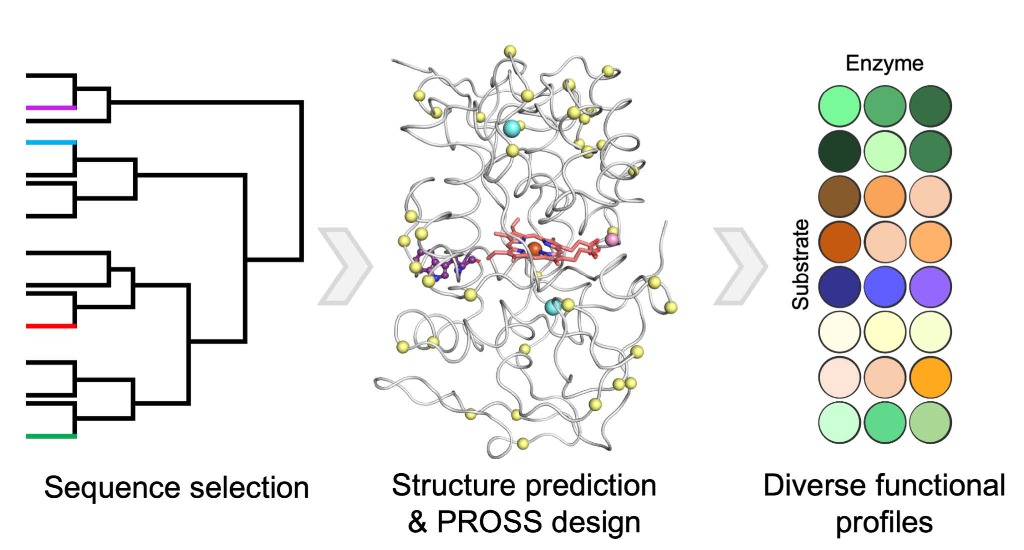
References:
Goldenzweig, A.; Goldsmith, M.; Hill, S. E.; Gertman, O.; Laurino, P.; Ashani, Y.; Dym, O.; Unger, T.; Albeck, S.; Prilusky, J.; Lieberman, R. L.; Aharoni, A.; Silman, I.; Sussman, J. L.; Tawfik, D. S.; Fleishman, S. J. Automated Structure- and Sequence-
Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63 (2), 337–346.
Khersonsky, O.; Lipsh, R.; Avizemer, Z.; Ashani, Y.; Goldsmith, M.; Leader, H.; Dym, O.; Rogotner, S.; Trudeau, D. L.; Prilusky, J.; Amengual-Rigo, P.; Guallar, V.; Tawfik, D. S.; Fleishman, S. J. Automated Design of Efficient and Functionally Diverse Enzyme Repertoires. Mol. Cell 2018, 72 (1), 178–186.e5.
Barber-Zucker et al Stable and functionally diverse versatile peroxidases designed directly from sequence bioRxiv
